We developed ISoLDE (Integrative Statistics of alleLe Dependent Expression), a novel non-parametric statistical method that directly infers allelic imbalance from RNA-seq data. ISoLDE learns the distribution of a specifically designed test statistic from the data and calls genes allelically imbalanced, bi-allelically expressed or undetermined. ISoLDE is available as a Bioconductor package.
We developed ISoLDE (Integrative Statistics of alleLe Dependent Expression), a novel non-parametric statistical method that directly infers allelic imbalance from RNA-seq data. ISoLDE learns the distribution of a specifically designed test statistic from the data and calls genes allelically imbalanced, bi-allelically expressed or undetermined. ISoLDE is available as a Bioconductor package.
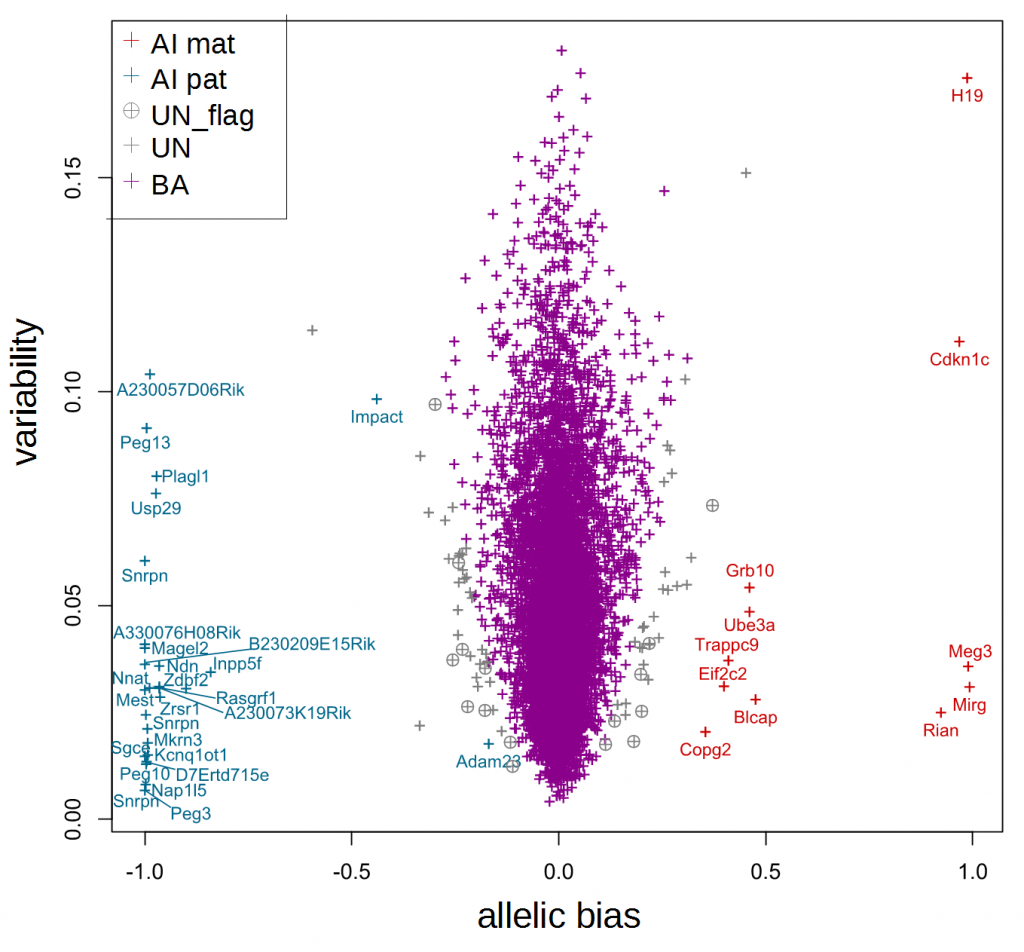
Output of the resampling version of ISoLDE. For each gene, the variability (denominator value of the Sg statistic) was plotted against the allelic bias (numerator value of the Sg statistic). Violet crosses correspond to bi-allelically expressed (‘BA’) genes. Red and blue crosses correspond to genes called maternally and paternally imbalanced (‘AI mat’ and ‘AI pat’, respectively). Grey crosses correspond to undetermined (‘UN’) genes. Grey circled crosses correspond to flagged genes (consistency or significance flag, ‘UN_flag’).
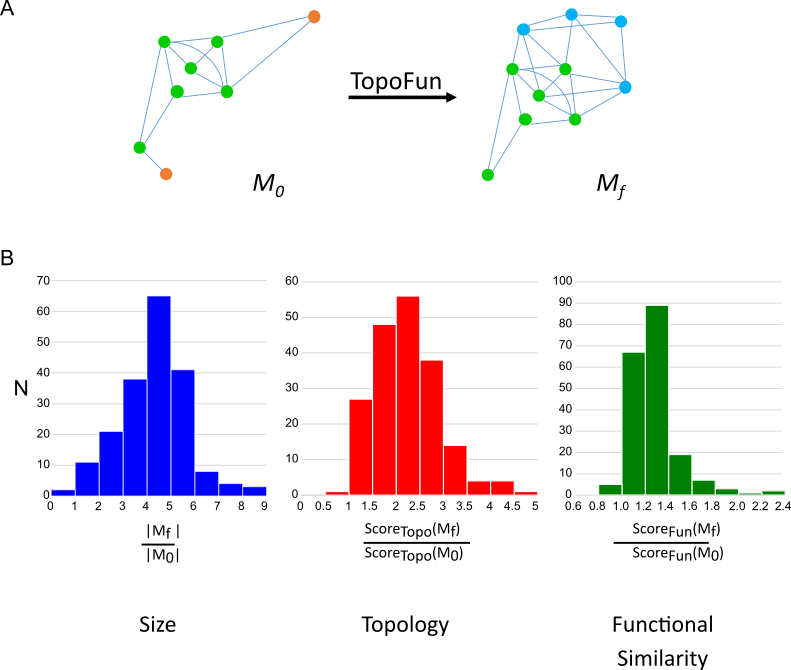
We also developped TopoFun, a novel machine learning method to identify functional modules in gene co-expression networks and complement Gene Ontology annotations.
A comprehensive, accurate functional annotation of genes is key to systems-level approaches. Forward and reverse genetics produced a substantial amount of data on gene functions; yet, a large fraction of genes are still poorly annotated, even in model organisms. One possible approach to complement existing annotations is to analyze gene co-expression as functionally related genes tend to be co-expressed.
Gene co-expression data are represented as high-dimensional graphs in which nodes denote genes and edges denote co-expression. TopoFun is a machine learning method that combines topological and functional information on co-expression modules. We first selected topological descriptors of gene co-expression modules that discriminate modules made of functionally related genes and modules made of randomly selected genes. Using the selected topological descriptors, we constructed a database of functional and random modules and performed Linear Discriminant Analysis to predict the type of a module. Starting from a given Gene Ontology Biological Process (GO-BP), we used a genetic algorithm to find genes whose co-expression with the largest clique of the GO-BP suggests that they may be functionally related.